




Welcome to Zhigang Shuai Group !
Zhigang Shuai 帅志刚
 |
School of Science and Engineering Tel: +86-755-23519615 and Department of Chemistry Email: zgshuai AT tsinghua.edu.cn ORCID: 0000-0003-3867-2331 |
||||
Honours and Awards
|







| 1983 | B.Sc. in Physics from Sun Yat-Sen University (Guangzhou) | |
|
1986
|
M. Sc. in Solid State Physics, Wuhan University, Supervisor: Nian-Ning Huang (Wuhan) | |
|
1989
|
Ph.D. in Theoretical Physics, Fudan University, Supervisor: Xin Sun (Shanghai) | |
|
1990 - 2001
|
Postdoc, Senior Research Associate in Jean-Luc Brédas Lab, University of Mons (Belgium) | |
|
2002
|
Research Professor, Institute of Chemistry, The Chinese Academy of Sciences (Beijing) | |
|
2008
|
Professor, Department of Chemistry, Tsinghua University (Beijing) | |
| 2022 | XQ Deng Presidential Chair Professor, School of Science and Engineering, The Chinese University of Hong Kong, Shenzhen | |
Journal Editorials
- Associate Editor of Acta. Chim. Sin. (CCS, 2012-),J. Mater. Chem. A (RSC, 2016-2019), and Univ. Chem. (PKU, 2015-2017).
- Chairman of ChemRxiv Governing Board (2021).
- Editorial Board Member of: National Science Review (CAS, 2017-), J. Mater. Chem. C (RSC, 2014-), Theor. Chem. Acc. (Springer, 2010-), Sci China Chem. (CAS, 2008-2017, 2019-2022), Progress in Chemistry (CAS, 2009-), Science Bulletin (CAS, 2015-); Adv. Theory Simul. (2017-); Chemical Journal of Chinese Universities (2019 -); Research (Science Partner Journal) (2021-); Synthetic Metals (2021-); Aggregate (2024-2026).
- (International) Advisory Board Member of: Journal of Physical Chemistry (ACS, 2017-2022), ChemPhysChem (2019-2022), WIRES Comput Mol Sci (Wiley, 2015-), Chem. Phys. Lett. (Elsevier,2015-), Chem. Asian J. (Wiley, 2014-), Nanoscale (RSC, 2012-), Nanoscale Advances (RSC, 2018-).
Services
- Vice President of the International Academy of Quantum Molecular Science (2018-2020, 2021-2023)
- Vice President of the Chinese Chemical Society (2019-2022, 2023-2026) 中国化学会副理事长
- Elected Executive Council Member of the Chinese Chemical Society (2011-2014, 2015-2018, 2019-2022, 2023-2026) 中国化学会常务理事
- Deputy Secretary-General of the Chinese Chemical Society (2007-2010, 2011-2014, 2015-2018)中国化学会副秘书长
- Chairman of the Theoretical Chemistry Committee of the Chinese Chemical Society (2015-2018) 中国化学会理论化学委员会主任
- National Representative of the IUPAC Committee on Chemistry Education (2010-2017)
- Associate Member of the IUPAC Physical and Biophysical Chemistry Division (2018-2019)
- Titular Member of the IUPAC Physical and Biophysical Chemistry Division (2020-2021, 2022-2023)
- Elected Bureau Member of IUPAC (2022-2023)
- Elected to the Executive Committee of IUPAC (2022-2023)
- Elected to the Executive Board of IUPAC (2024-2025, 2026-2027)
- IUPAC Coordinator for the International Year of Basic Science for Sustainable Development (IYBSSD 2022), an initiative approved by United Nation General Assembly 2021
- Vice Chairman of the Academic Committee of the MOE Key Laboratory of Organic OptoElectronics and Molecular Engineering, Tsinghua University (2018-)
- Member of the Academic Committee of the following institutions: The Institute of Chemistry of the CAS (2002-2023), State Key Laboratory of Physical Chemistry of Solid Surface (Xiamen University, 2009-2022), CAS Key Laboratory of Organic Solids (2018-), The MOE Key Laboratory of Mesoscopic Chemistry (Nanjing University, 2008-), Department of Chemistry of Tsinghua University (2008-2023), The MOE Key Laboratory of Organosilicon Chemistry and Materials Technology (Hangzhou Normal University, 2016-), Tsinghua Institute for Flexible Electronics (2018-).
- Member of the following CCS committees: Theoretical Chemistry Committee (2007-), Organic Solids Committee (2002-), Physical Organic Chemistry Committee (2017-), Physical Chemistry Division (2019-)
- 中国科协国际科学理事会中国委员会(ISC-CHINA)委员 (2022-2025, 2025-2028).
Recent Selected papers
- Qian Peng, Yuanping Yi, Zhigang Shuai*, Jiushu Shao, “Toward Quantitative Prediction of Molecular Fluorescence Quantum Efficiency: Role of Duschinsky Rotation”, Journal of The American Chemical Society 2007, 129, 9333-9339.
- Yingli Niu, Qian Peng, Zhigang Shuai*, “Promoting-Mode Free Formalism for Excited State Radiationless Decay Process with Duschinsky Rotation Effect”, Science in China Series B: Chemistry 2008, 51, 1153-1158.
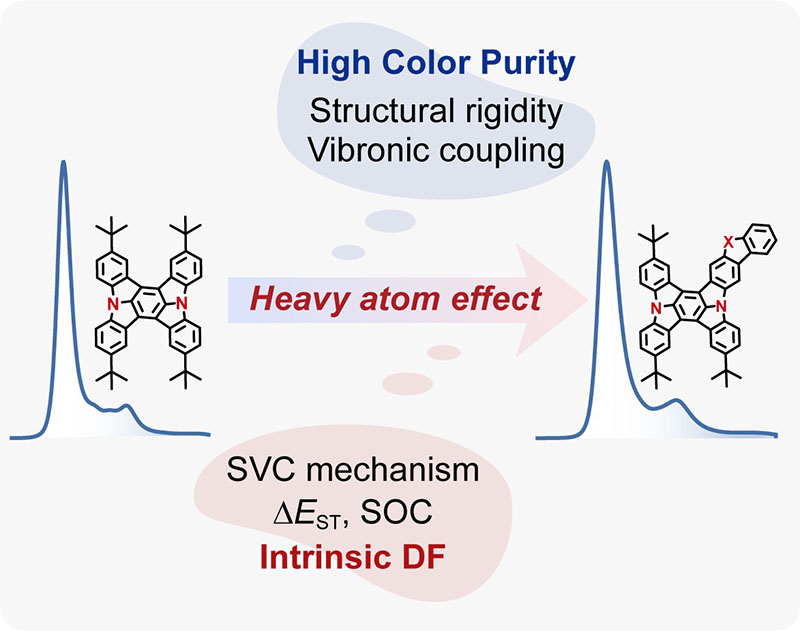
- Huili Ma, Qian Peng*, Zhongfu An, Wei Huang, Zhigang Shuai*, “Efficient and Long-Lived Room-Temperature Organic Phosphorescence: Theoretical Descriptors for Molecular Designs” , Journal of The American Chemical Society 2019, 141, 1010-1015.
- Zhigang Shuai*, “Thermal Vibration Correlation Function Formalism for Molecular Excited State Decay Rates”, Chinese Journal of Chemistry 2020, 38, 1223-1232.
- Qi Ou, Yihan Shao, Zhigang Shuai*, “Enhanced reverse intersystem crossing promoted by triplet exciton–photon coupling”, Journal of The American Chemical Society 2021, 143, 17786-17792.
- Qian Peng, Huili Ma, Zhigang Shuai*, “Theory of long-lived room-temperature phosphorescence in organic aggregates”, Accounts of Chemical Research 2021, 54, 940-949.
- Qi Ou, Qian Peng, Zhigang Shuai*, “Computational Screen-out Strategy for Electrically Pumped Organic Laser Matetials”, Nature Communications 2020, 11, 4485.
- Zhigang Shuai, “Faster Holes by Delocalization”, Nature Materials 2023, 22, 1277-1278.
- Guangjun Nan, Xiaodi Yang, Linjun Wang, Zhigang Shuai*, Yi Zhao, “Nuclear Tunneling Effects of Charge Transport in Rubrene, Tetracene, and Pentacene”, Physical Review B 2009, 79, 115203~1-9.
- Mengqiu Long, Ling Tang, Dong Wang, Linjun Wang, Zhigang Shuai*, “Theoretical Predictions of Size-Dependent Carrier Mobility and Polarity in Graphene”, Journal of the American Chemical Society 2009, 131, 17728-17729.
- Mengqiu Long, Ling Tang, Dong Wang, Yuliang Li, Zhigang Shuai*, “Electronic Structure and Carrier Mobility in Graphdiyne Sheet and Nanoribbons: Theoretical Predictions”, ACS Nano 2011, 5, 2593-2600.
- Wen Shi, Tianqi Zhao, Jinyang Xi, Dong Wang*, Zhigang Shuai*, “Unravelling Doping Effects on PEDOT at the Molecular Level: From Geometry to Thermoelectric Transport Properties”, Journal of the American Chemical Society 2015, 137, 12929 – 12938.
- Hua Geng, Qian Peng, Linjun Wang, Haijiao Li, Yi Liao, Zhiying Ma, Zhigang Shuai*, “Towards Quantitative Prediction of Charge Mobility in Organic Semiconductors: Tunneling Enabled Hopping Model”, Advanced Materials 2012, 24, 3568 – 3572.
- Zhigang Shuai*, Linjun Wang, Qikai Li, “Evaluation of Charge Mobility in Organic Materials: From Localized to Delocalized Description at First-Principles Level”, Advanced Materials 2011, 23, 1145-1153.
- Weitang Li, Jiajun Ren, Zhigang Shuai*, “A General Charge Transport Picture for Organic Semiconductors with Nonlocal Electron-Phonon Couplings”, Nature Communications 2021, 12, 4260.
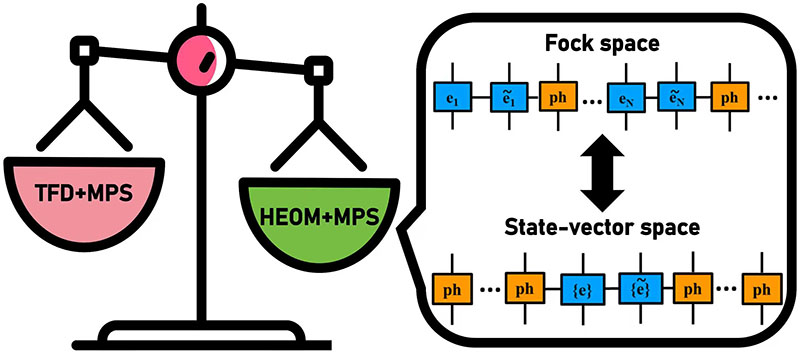
- Jiajun Ren, Zhigang Shuai*, Garnet Kin-Lic Chan*, “Time-dependent Density Matrix Renormalization Group Algorithm for Nearly Exact Absorption and Fluorescence Spectra of Molecular Aggregates at Both Zero and Finite Temperature”, Journal of Chemical Theory and Computation 2018, 14, 5027-5029.
- Yuanheng Wang, Jiajun Ren*, Zhigang Shuai*, “Minimizing Non-radiative Decay in Molecular Aggregate Through Control of Excitonic Coupling”, Nature Communications 2023, 14, 5056.
- Tong Jiang, Weitang Li, Jiajun Ren*, Zhigang Shuai*, “Finite Temperature Dynamical Density Matrix Renormalization Group for Spectroscopy in Frequency Domain”, Journal of Physical Chemistry Letters 2020, 11, 3761-3768.
- Tong Jiang, Jiajun Ren, Zhigang Shuai*, “Unified Definition of Exciton Coherence Length for Exciton-Phonon Coupled Molecular Aggregates”, Journal of Physical Chemistry Letters 2023, 14, 4541-4547.
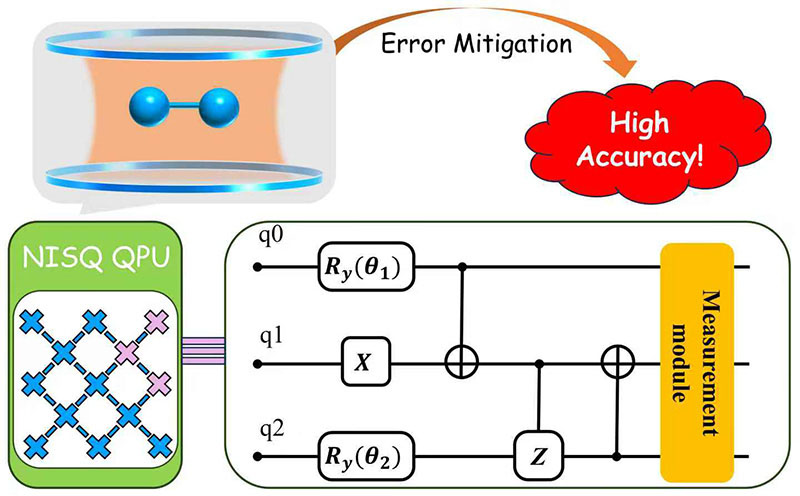
- Weitang Li, Jiajun Ren, Sainan Huai, Tianqi Cai, Zhigang Shuai*, Shengyu Zhang*, “Efficient Quantum Simulation of Electron-Phonon Systems by Variational Basis State Encoder”, Physical Review Research 2023, 5, 023046.
Please visit the web page Publications for the full list.
Developing and applying new theoretical and computational methods
to study optoelectronic functional materials
The research in the Shuai Group involves theoretical and computational studies of phenomena in organic and polymeric functional materials.
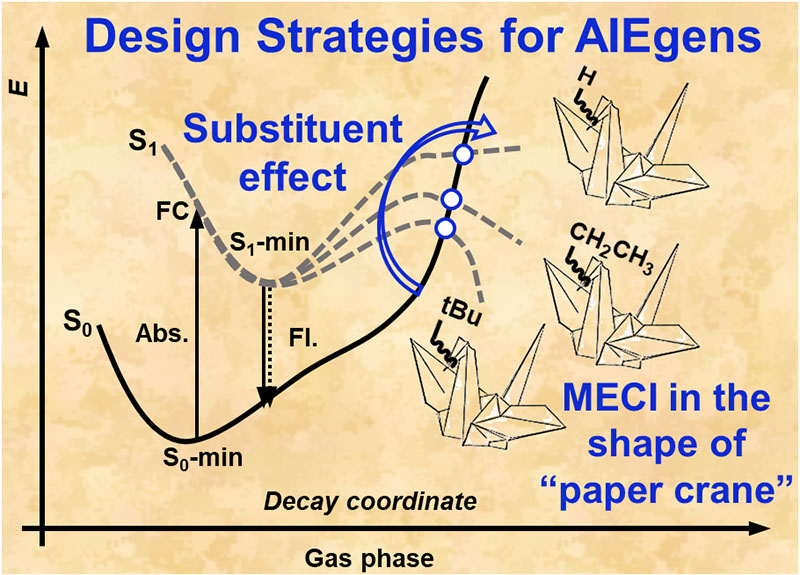
A primary goal is to develop and apply new computational methods to explain and predict the opto-electronic functions of organic functional materials. For instance, we are interested in the charge/thermoelectric transports in organic semiconductors and carbon nano-ribbon and sheets, electronic excited state decay processes relevant to organic light-emitting quantum efficiency and spectrum, charge and exciton dynamics in polymer photovoltaics, thermoelectric figure of merits, nonlinear optical responses and multiphoton absorption, supramolecular self-assembly and disassembly.
These also include methods for correlated electron such as quantum chemistry density matrix renormalization group and equation of motion coupled cluster theory, both at semiempirical levels.