2026 News
|
2026/06/15
|
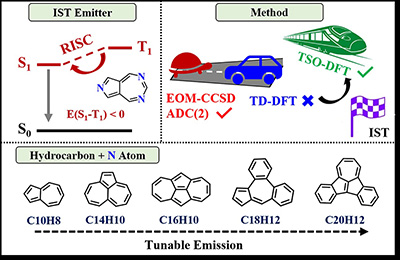
Congratulations to Dr Ramalingam Mahaan for the new paper accepted for publication in JPCL! Hund’s rule, derived from direct exchange (K), dictates that triplet (T1) lies below singlet (S1), leading to the 25% limit for electrofluorescence. Kinetic exchange (−t2/U) from charge-transfer perturbation can stabilize S1 over T1, enabling inverted singlet–triplet (IST) gaps and offering a promising route to triplet harvesting. Such materials are exceedingly rare, and systematic design remains elusive. Here, we employ newly developed target-state optimized density functional theory (TSO-DFT) for cost-effective excited-state calculations, yielding reliable IST signatures with the B3LYP functional. Subsequently, we apply a high-throughput virtual screening (HTVS) to explore nitrogen-doped hydrocarbons derived from C10H8, C14H10, C16H10, C18H12, and C20H12 molecular cores via isomerization. This workflow screens 118 081 viable structures down to 34 potential candidates exhibiting negative S1–T1 gaps, an appreciable oscillator strength of ≥0.01, and fast RISC rates (∼108 s–1). This study provides a discovery pipeline for novel IST emitters and a curated library of visible-light candidates for next-generation optoelectronics. |
|
2026/06/15
|
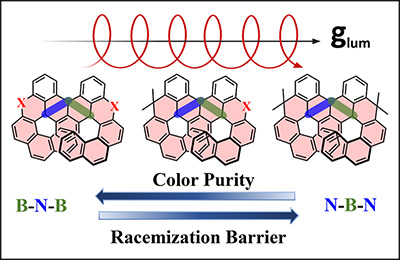
Congratulations to Dr Ramalingam Mahaan for the new paper accepted for publication in JPCC! Circularly polarized multiple-resonance thermally activated delayed fluorescence (CP-MR-TADF) emitters have gained immense interest in advanced optoelectronics due to their unique combination of narrow emission, efficient TADF, and chirality. However, their rational design requires a deeper understanding of the intricate structure-property relationships that govern their photophysical and chiroptical behavior. In this work, we systematically explore a series of helicene-based CP-MR-TADF emitters through four strategic modifications: the core framework (B-N-B vs. N-B-N), MR-TADF skeleton architecture, chiral unit variation, and the heavy-atom effect (O, S, Se). Employing high-level STEOM-DLPNO-CCSD calculations benchmarked against experimental data, we theoretically design 28 novel molecules to target highly efficient CPL properties and accurately predict excited-state energies. Our analysis reveals that the B-N-B framework consistently yields superior color purity, with FWHM values as narrow as ∼14 nm, resulting from minimized structural relaxation. The incorporation of heavy atoms (O, S, Se) systematically enhances reverse intersystem crossing (RISC) rates up to ∼107 s−1 by strengthening SOC and reducing the ΔEST gap, making it ideally suited for the TADF process. All designed molecules exhibit high luminescence dissymmetry factors on the order of ∼10−3, with the N-B-N framework yielding the maximum values, up to 4.17×10−3, attributed to its characteristic enhanced transition electric-magnetic dipole moment angles. Notably, the N-B-N framework significantly increases the racemization barrier by ∼20 kcal mol−1 relative to the B-N-B framework, thereby ensuring configurational stability under ambient conditions. This study establishes clear structure-property relationships and outlines a design framework for developing high-performance CP-MR-TADF emitters with balanced narrow-width emission, efficient triplet harvesting, and strong chiroptical activity. |
|
2026/05/25
|
Congratulations to Wenjie Zhang!
Congratulations to Wenjie Zhang for completing his PhD Dissertation Defense!
|

2025 News
|
2025/12/19
|
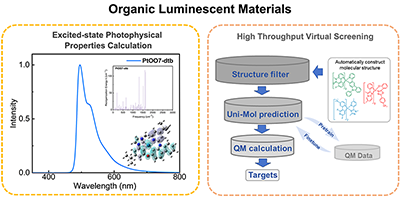
Congratulations to Dr Rongrong Li for the new paper accepted for publication in CSR! The theoretical design of highly efficient, low roll-off and full-color emission organic materials is of great interest, although there are great challenges due to the limitations of the present-day methodology. In this review, we present progress achieved in our group on the theoretical and computational investigation for the structure–property relationships and screening strategy for organic fluorescent molecules, selection of thermally activated delayed fluorescence (TADF) and multi-resonance TADF (MR-TADF) molecules for optically and electrically pumped lasing application, and high-throughput virtual screening of phosphorescent organometallic complexes. We combined a quantum chemistry method with the molecular representation learning model Uni-Mol and rate theory-based molecular material property prediction package (MOMAP) developed in our group. Finally, we outline the limitation of current computational protocols and the future directions for organic luminescent materials. |
||
|
2025/12/19
|
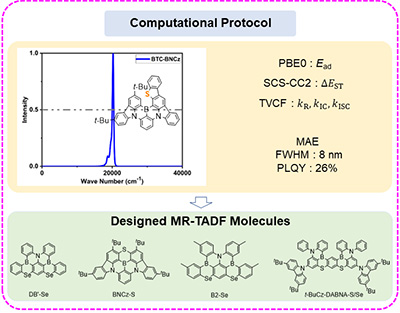
Congratulations to Dr Rongrong Li for the new paper accepted for publication in JCP! Full width at half maximum (FWHM) is an important indicator for color purity in molecular optical emission. In addition, brightness is determined by photoluminescence quantum efficiency (PLQY). Thermally activated delayed fluorescence (TADF) can convert the electro-pumped triplet states into emissive singlets. Especially, recent experiments suggest that multiple resonance TADF molecules doped with heavy atoms S/Se could effectively avoid the efficiency roll-off by promoting the reverse intersystem crossing (RISC) process. We propose a computational protocol to evaluate FWHM and PLQY based on quantum chemistry calculations and thermal vibration correlation function formalism, which is of great potential to design highly efficient and color-pure molecules. We further build a robust correlation between the reorganization energy of emission state λemS1S0 with FWHM and the inverse of reorganization energy of the intersystem crossing (ISC) process 1/λS1T1 with experimental reverse ISC rate constant kexpRISC, crucial for TADF. Our computational method and findings can be used for the molecular design of organic light-emitting diode materials. |
||
|
2025/12/10
|
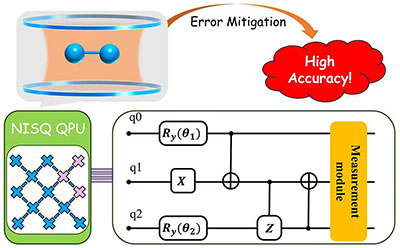
Congratulations to Xinyan Jiang for the new paper accepted for publication in JCP! Quantum computing on near-term noisy intermediate-scale quantum devices holds significant promise for simulating complex chemical systems. Among various variational quantum algorithms, the adaptive derivative-assembled pseudo-Trotter ansatz variational quantum eigensolver (ADAPT-VQE) is widely used for generating molecule-specific adaptive ansätze for different molecules, yet its measurement requirement is extensive, calling for suitable optimizers. In this study, we utilize the ADAPT-VQE algorithm enhanced by a powerful optimizer termed sequential optimization with an approximate parabola (SOAP) to calculate molecular energies. These computations are carried out through classical simulations using the TenCirChem software. Our results demonstrate the efficiency and robustness of the SOAP optimizer for ADAPT-VQE. Furthermore, we show that SOAP performs effectively across different ADAPT-VQE ansatz element pools. This work presents a strategy to mitigate the substantial measurement requirements associated with ADAPT-VQE. |
||
|
2025/10/28
|
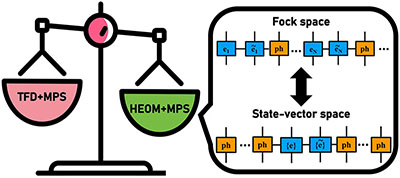
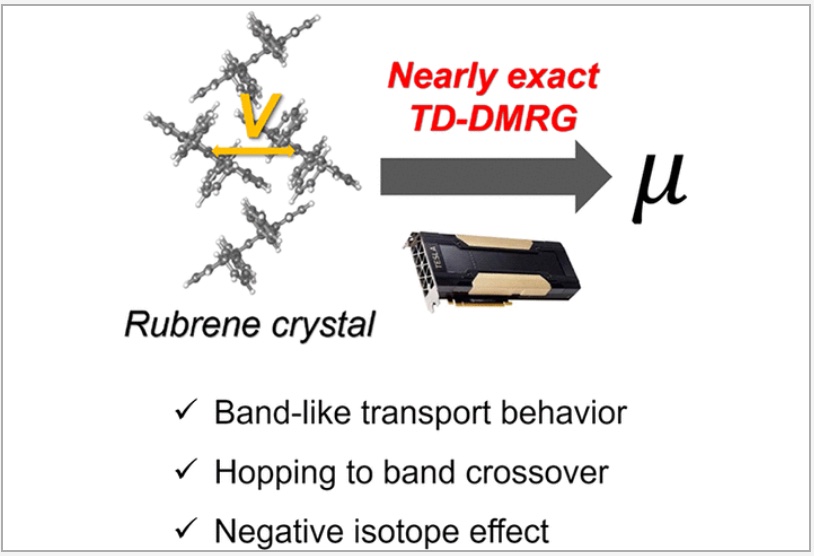
Congratulations to Hengrui Yang for the new paper accepted for publication in JCTC! We present a Hierarchical Equations of Motion(HEOM) approach in the Matrix Product State (MPS) formalism to simulate carrier transport in molecular aggregates described by anelectron−phonon Hamiltonian with bosonic dissipation. Transport properties are evaluated through time-dependent population analysis and mobility calculations. The method’s validity is rigorously established through benchmarking against conventional HEOM.Comparative analysis with Thermo-Field Dynamics combined with MPS (TFD + MPS) reveals fundamental similarities and differences in their effective Hamiltonians and demonstrates the superior accuracy and computational efficiency of our HEOM + MPS framework. For single-electron systems, we introduce state-vector space configurations that enhance performance beyond traditional Fock space approaches. Results confirm that our method provides a robust, nearly exact, and efficient numerical quantum dynamic approach for carrier transport in dissipative bosonic environments. |
||
|
2025/10/25
|
Congratulations to Hengrui Yang!
Congratulations to Hengrui Yang for completing his PhD Dissertation Defense!
|
||
|
2025/08/14
|
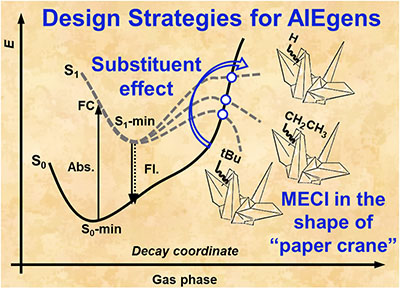
Congratulations to Ping-An Yin for the new paper accepted for publication in JCTC! Aggregation-induced emission (AIE) has become a key focus in luminescent material development, with substituent modulation being a critical strategy for expanding AIE systems. The S1/S0 minimum energy conical intersection (MECI) significantly influences molecular photophysical properties, making it essential for understanding the AIE phenomenon. Here, we employ anthracene derivatives, known for their chemical versatility and applications in organic light-emitting diodes (OLEDs), to systematically investigate the effects of substituents on the S1/S0-MECI. We select 22 anthracene derivatives with varied electron-donating and electron-withdrawing substituents and explore their impacts on the S1/S0-MECI relative energy and molecular structure. Our findings reveal that strong electron-donating or electron-withdrawing groups at the C9-position effectively lower the S1/S0-MECI relative energy of the gaseous phase singly substituted anthracene derivatives, thus enhancing the AIE phenomenon of such molecules. Additionally, doubly substituted derivatives on the same ring also slightly reduce the S1/S0-MECI relative energy of the isolated molecule. Based on these insights, we propose a novel AIE molecular design strategy focusing on modulating S1/S0-MECI through strategic substituent selection, leading to the identification of 24 AIEgens candidates among 81 anthracene derivatives. In summary, our study provides a systematic approach to designing AIE molecules by modulating the S1/S0-MECI through a substituent effect. The validity of this strategy is confirmed using the 9-tBu-Ant molecule with quantitative calculations. |
||
|
2025/07/30
|
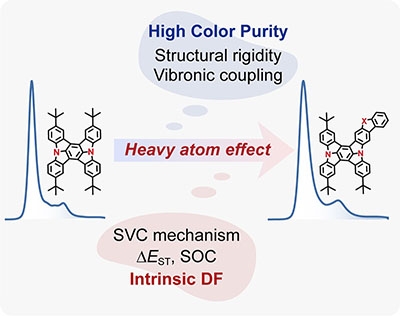
Congratulations to Yaxin Wang for the new paper accepted for publication in JPCL! Indolo[3,2,1-jk] carbazole (ICz)-based multiple resonance (MR) organic emitters bearing small full width at half-maximum (fwhm) values are of interest in organic light-emitting diodes. The challenge is to boost the intrinsic delayed fluorescence by enhancing the reverse intersystem crossing (RISC) process to avoid efficiency roll-off. Here, we propose the utilization of the nonmetallic heavy-atom effects to promote the RISC process while keeping the advantage of structural rigidity for narrowband emission. The emission spectrum and quantum efficiency were investigated by using quantum chemistry calculations coupled with our thermal vibration correlation function (TVCF) formalism. For heavy-atom embedded dICz-MR emitters, the synergistic effect of spin−vibronic coupling and spin−orbit coupling promotes the potential RISC channels (T1 → T2/T3 → S1), the effective kRISC, up to 106 s−1. Besides, heavy-atom embedded para-linked ICz-MR emitters have little effect on emission width due to the considerable structural rigidity and low-frequency dominated vibronic coupling. |
||
|
2025/07/07
|
|
||
|
2025/05/12
|
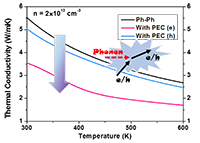
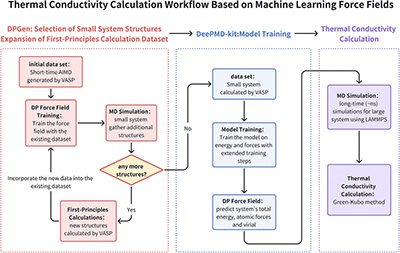
Congratulations to Wenjie Zhang for the new paper accepted for publication in Phys. Chem. Chem. Phys.! The accurate prediction of lattice thermal conductivity in organometallic thermoelectric materials is crucial for advancing energy conversion technologies. Methods based on molecular dynamics simulations can solve this problem well, but require force fields with sufficiently high accuracy. Due to the complexity of chemical bonding in organometallic complex materials, the development of force fields with high predictivity has been a long standing challenge, particularly when thermal transport is concerned which requires even greater accuracy. In recent years, the rapid advancement of machine learning force fields has offered substantial potential for addressing these issues. However, there remain challenges for materials with large organometallic complexes in one unit cell and both inter- and intramolecular interactions. In this work, we employ an active learning approach combined with deep neural networks to develop a force field taking copper phthalocyanine as an example. The model utilizes a local environment descriptor for representation without explicitly characterizing the metal–organic coordination. The nonlinear mapping capabilities of deep neural networks enable the model to effectively capture higher-order many-body interactions. Furthermore, we utilized the Green–Kubo method to calculate the thermal conductivity of copper phthalocyanine, revealing a value of 0.49 W m-1 K-1 at 300 K, consistent with experimental findings (0.39 W m-1 K-1). This result significantly surpasses previous work with classical force fields. This work represents a significant advancement in demonstrating that machine-learning force fields can effectively characterize interactions in metal–organic complex systems and can significantly advance the development and discovery of organometallic thermoelectric materials. |
||
|
2025/05/08
|
Congratulations to Pingan Yin!
Congratulations to Pingan Yin for completing his PhD Dissertation Defense!
|
||
|
2025/04/07 近五年榜单:
|
|||
|
2025/03/18
|
Congratulations to Xiao Chen for the new paper accepted for publication in J. Phys. Chem. A! Heavy metal complexes are important organic lightemitting diode (OLED) materials, with the advantage of theoretically up to 100% quantum efficiency, but suffer from low color purity, specifically due to a large emission spectral width. Recently, several tetradentate platinum(II) complexes have been found to demonstrate a narrow emission width. In order to suggest a molecular design strategy to achieve narrow emission width, we combine density functional theory (DFT) and its time-dependent formalism coupled with the thermal vibrational correlation function (TVCF) formalism to evaluate the emission energy and spectral line shape for 50 tetradentate platinum(II) complex compounds. We have benchmarked the computational approach by testing 14 xc-functionals and 4 potential energy surface models. We find that the pyridinecarbazole-oxygen structure is the essential moiety to achieve high color purity for the Pt(II) coordination compounds, which can suppress the excitation of low-frequency vibrational relaxation motion during phosphorescence emission, leading to a narrow emission spectrum. |
||
|
2024/10/29 MOMAP Workshop 2024 |
|||
|
2024/09/21
|
The MOMAP Workshop 2024 is to be held in Changsha, China from Oct 24 to 27, 2024. Please click the following links to visit the MOMAP Workshop 2024 websites for more details: |
||
|
2024/09/02
|
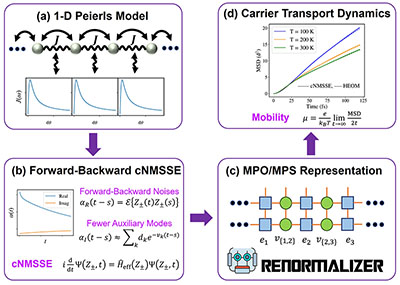
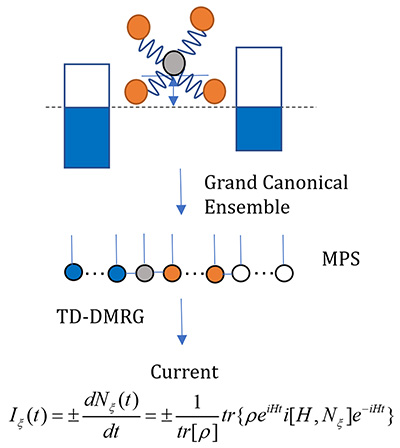
Congratulations to Liqi Zhou for the new paper accepted for publication in J. Chem. Phys.! Evaluation of the charge transport property of organic semiconductors requires exact quantum dynamics simulation of large systems. We present a numerically nearly exact approach to investigate carrier transport dynamics in organic semiconductors by extending the non-Markovian stochastic Schrödinger equation with complex frequency modes to a forward–backward scheme and by solving it using the matrix product state (MPS) approach. By utilizing the forward–backward formalism for noise generation, the bath correlation function can be effectively treated as a temperature-independent imaginary part, enabling a more accurate decomposition with fewer complex frequency modes. Using this approach, we study the carrier transport and mobility in the one-dimensional Peierls model, where the nonlocal electron–phonon interaction is taken into account. The reliability of this approach was validated by comparing carrier diffusion motion with those obtained from the hierarchical equations of motion method across various parameter regimes of the phonon bath. The efficiency was demonstrated by the modest virtual bond dimensions of MPS and the low scaling of the computational time with the system size. |
||
|
2024/08/02 |
Congratulations to Shanhao Deng for the new paper accepted for publication in ChemPhotoChem! In microcavity, strong coupling between light and molecules leads to the formation of hybrid excitations, i.e., the polaritons, or exciton-polaritons. Such coupling may alter the energy landscape of the system and the optical properties of the material, making it an effective approach for controlling the light emission from molecular materials. However, due to the complexity of vibrational modes, spectroscopic calculations for organic exciton-polaritons remain to be challenging. In this work, based on the linear-response quantum-electrodynamical time-dependent density functional theory (QEDTDDFT), we employ the thermal vibrational correlation function (TVCF) formalism to calculate the molecular optical spectrum of the lower polaritons (LP) at first-principles level for three molecules, i.e., anthracene, distyrylbenzenes (DSB), and rubrene. The polaron decoupling effect is confirmed from our first-principles computations. The theoretical emission spectra of LP provide new insights for aiding molecular and device design in microcavities that are otherwise hindered due to the lack of vibrational information. |
||
|
2024/08/02
|
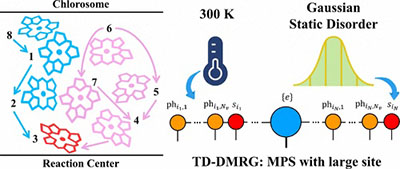
Congratulations to Zirui Sheng for the new paper accepted for publication in JCTC! Photosynthesis is a fundamental process that converts solar energy into chemical energy. Understanding the microscopic mechanisms of energy transfer in photosynthetic systems is crucial for the development of novel optoelectronic materials. Simulating these processes poses significant challenges due to the intricate interactions between electrons and phonons, compounded by static disorder. In this work, we present a numerically nearly exact study using the time-dependent density matrix renormalization group (TD-DMRG) method to simulate the quantum dynamics of the Fenna-Matthews-Olson (FMO) complex considering an eight-site model with both thermal and static disorders. We employ the thermo-field dynamics formalism for temperature effects. We merge all electronic interactions into one large matrix product state (MPS) site, boosting accuracy efficiently without increasing complexity. Previous combined experimental and computational studies indicated that the static disorders range from 30 to 90 cm-1 for different FMO sites. We employ a Gaussian distribution and the auxiliary bosonic operator approach to consider the static disorder in our TD-DMRG algorithm. We investigate the impact of different initial excitation sites, temperatures, and degrees of static disorder on the exciton dynamics and temporal coherence. It is found that under the influence of the experimentally determined static disorder strength, the exciton population evolution shows a non-negligible difference at zero temperature, while it is hardly affected at room temperature. |
||
|
2024/07/04
|
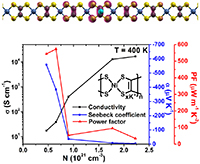
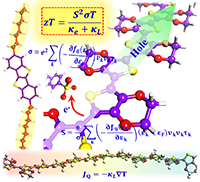
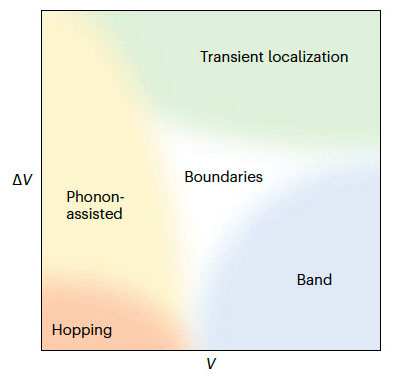
Congratulations to Dr Yufei Ge for the new paper accepted for publication in Phys. Rev. B! Organic molecular materials are potential high-performance thermoelectric materials. Theoretical understanding of thermoelectric conversion in organic materials is essential for rational molecular design for efficient energy conversion materials. In organic materials, nonlocal electron-phonon coupling plays a vital role in charge transport and leads to complex transport mechanisms, including hopping, phonon assisted, band, and transient localization. In this work, based on the time-dependent density matrix renormalization group method, we look at the role of nonlocal electron-phonon coupling on the thermoelectric conversion in organic systems described by the Holstein-Peierls model. We calculate the current-current correlation and the heat current-current correlation functions. We find that (i) nonlocal electron-phonon coupling has a very weak influence on the Seebeck coefficient because of the cancellation between the heat current-current correlation function and the current-current correlation function, but it has a strong influence on the conductivity through dynamic disorders; and (ii) doping concentration has a strong influence on both the conductivity and Seebeck coefficient, and the optimal doping ratio to reach the highest power factor is 3%–10% fillings when the Holstein-Peierls model is valid. These findings suggest that we can design organic materials with higher power factors by first enhancing mobility through rational design, and then searching for the optimal doping ratio. |
||
|
2024/05/13
|
Congratulations to Yufei Ge!
Congratulations to Yufei Ge for completing his PhD Dissertation Defense!
|
||
|
2024/04/15
|
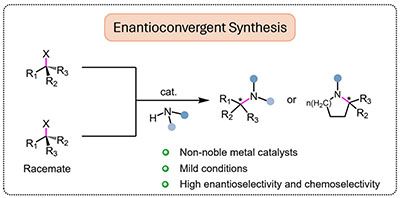
Congratulations to Dr Rongrong Li for the new paper accepted for publication in ChemCatChem! Nitrogen containing compounds like chiral amines have been widely used in synthetic medicines, biomass, and fine chemical products because of their particularly valuable motifs and exceedingly broad range of functionality. Enantioconvergent synthesis of chiral amines has high atomic economy and enantioselectivity with theoretically 100 % yield of enantioenriched products. In this concept article, we hope to provide a further understanding of chiral amines enantioconvergent synthesis methodology with an introduction of recent progresses in enantioconvergent amination reactions catalyzed by early transition metals and facilitate the development of enantioconvergent C(sp3)−N coupling for benign and efficient preparation of highly valuable chiral nitrogen-containing units. |
||
|
2023 News |
|||
|
2023/11/30
|
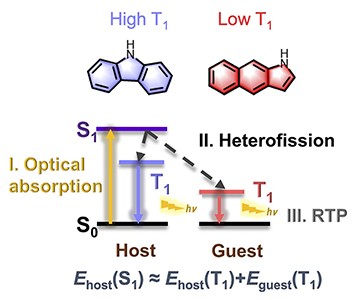
Congratulations to Dr Qi Sun for the new paper accepted for publication in Adv. Optical Mater.! Room temperature phosphorescence (RTP) from pure organic materials, whether in crystalline or film phases, has recently attracted considerable attention. Experimental evidence increasingly suggests that RTP originates from isomeric dopants (impurity) rather than pure compounds. The underlying mechanism and molecular design principles have remained elusive. Herein, the “heterofission mechanism” for RTP is proposed, wherein a singlet excited state is split into two triplets, one remains within the host, while the other migrates to the guest (dopant) molecule, satisfying Ehost(S1) ≈ Ehost(T1) + Eguest(T1). It is found that all the dopants possess a low triplet excited state, meeting the energy requirement for the heterofission process. The sum of the calculated emission spectra from these two triplets overlaps well with the experimentally measured broad phosphorescent spectra. Furthermore, the calculated heterofission rates are expected to occur at the picosecond timescale. Efficient Dexter energy transfer leads to the guest predominantly dominating the RTP. Based on this mechanism, we can predict potential host and guest candidates to expand the family of pure organic RTP materials. |
||
|
2023/11/30
|
Congratulations to Prof. Zhigang Shuai for the new paper accepted for publication in Nature Materials! Terahertz photoconductivity measurements coupled with theoretical modelling reveals that thermal transient excitations to more delocalized states enhances hole mobility in organic molecular semiconductors. |
||
|
2023/9/20
|
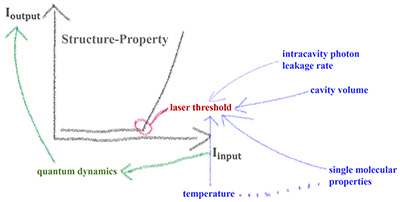
Congratulations to Dr Bin Zhang for the new paper accepted for publication in JPCL! The quantum dynamic (QD) study of organic lasing (OL) is a challenging issue in organic optoelectronics. Previously, the phenomenological method has achieved success in describing experimental observation. However, it cannot directly bridge the laser threshold (LT) with microscopic parameters, which is the advantage of the QD method. In this paper, we propose a microscopic OL model and apply time-dependent wave packet diffusion to reveal the microscopic QD process of optically pumped lasing. LT is obtained from the onset of output as a function of optical input pumping. We predict that the LT has an optimal value as a function of the cavity volume and depends linearly on the intracavity photon leakage rate. The calculated structure−property relationships between molecular parameters and the LT are in qualitative agreement with the experimental results, confirming the reliability of our approach. This work is beneficial for understanding the OL mechanism and optimizing the design of organic laser materials. |
||
|
2023/9/7
|
Congratulations to Hengrui Yang for the new paper accepted for publication in JCTC! Quantum transport in molecular junctions has attracted great attention. The charge motion in a molecular junction can cause geometric deformation, leading to strong electron phonon coupling, which was often overlooked. We have formulated a nearly exact method to assess the time-dependent current and occupation number in the molecular junction modeled by the electron−phonon coupled bridge state using the time-dependent density matrix renormalization group (TD-DMRG) method. The oscillation period and amplitude of the current are found to be dependent on the electron phonon coupling strength and energy level alignment with the electrodes. In an attempt to better understand these phenomena, we have devised a new approximation that explains the bistability phenomenon and the behavior of steady currents in the strong electron−phonon coupling regime. Comparisons have been made with the multilayermulticonfiguration time-dependent Hartree (ML-MCTDH) method and the analytical result in the purely electronic limit. Furthermore, we explore the entropy of different orderings, extending to the electron phonon model problems. Regarding finite temperature, the thermal Bogoliubov transformation of both fermions and bosons is used and compared with imaginary time evolution results. |
||
|
2023/8/23
|
|||
|
2023/8/23
|
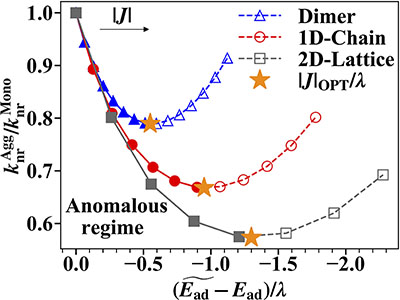
Congratulations to Yuanheng Wang for the new paper accepted for publication in Nature Communications! The widely known “Energy Gap Law” (EGL) predicts a monotonically exponential increase in the non-radiative decay rate (knr) as the energy gap narrows, which hinders the development of near-infrared (NIR) emissive molecular materials. Recently, several experiments proposed that the exciton delocalization in molecular aggregates could counteract EGL to facilitate NIR emission. In this work, the nearly exact time-dependent density matrix renormalization group (TD-DMRG) method is developed to evaluate the non-radiative decay rate for exciton-phonon coupled molecular aggregates. Systematical numerical simulations show, by increasing the excitonic coupling, knr will first decrease, then reach a minimum, and finally start to increase to follow EGL, which is an overall result of two opposite effects of a smaller energy gap and a smaller effective electron-phonon coupling. This anomalous non-monotonic behavior is found robust in a number of models, including dimer, one-dimensional chain, and two-dimensional square lattice. The optimal excitonic coupling strength that gives the minimum knr is about half of the monomer reorganization energy and is also influenced by system size, dimensionality, and temperature. |
||
|
2023/7/28
|
Congratulations to Prof Zhigang Shuai!
  ...
|
||
|
2023/7/3
|
Congratulations to Tong Jiang!
Congratulations to Tong Jiang for obtaining the excellent Ph.D. thesis!
|
||
|
2023/5/23
|
Congratulations to Yuanheng Wang!
Congratulations to Yuanheng Wang for completing his PhD Dissertation Defense!
|
||
|
2023/5/23
|
Congratulations to Tong Jiang!
Congratulations to Tong Jiang for completing his PhD Dissertation Defense!
|
||
|
2023/5/23
|
Congratulations to Qi Sun!
Congratulations to Qi Sun for completing her PhD Dissertation Defense!
|
||
|
2023/5/10
|
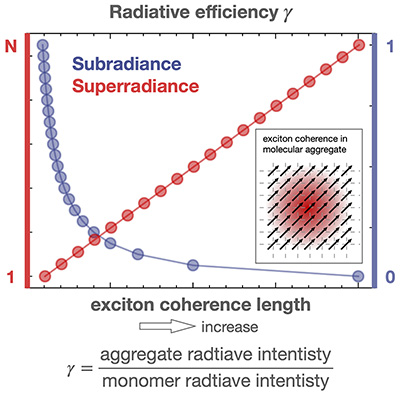
Congratulations to Tong Jiang for the new paper accepted for publication in J. Phys. Chem. Lett.! Exciton coherence length (ECL) characterizes the spatial extent of coherently delocalized excited states of molecular aggregates. Constructive/destructive superpositions of coherent molecular dipoles lead to superradiance/subradiance, where the radiative rate is enhanced/suppressed compared to that of a single molecule. Longer ECLs correspond to faster/slower radiative rates for superradiant/subradiant aggregates. However, previous ECL definitions fail to produce monotonic relationships when exciton−phonon coupling is considered, even for simple 1D exciton−phonon systems. This problem is exacerbated for 2D aggregates with both constructive and destructive superpositions. In this Letter, we propose a novel ECL definition by virtue of sum rule for oscillator strengths, ensuring a bijective and monotonic relationship between ECL and radiative rate for both 1D/2D superradiant and subradiant aggregates. Using numerically accurate time-dependent matrix product states, we study large-scale, exciton−phonon coupled 2D aggregates and predict the existence of maximum superradiance at finite temperature, in contrast to the previously believed 1/T law. Our results provide new insights into the design and optimization of efficient light emitting materials.
|
||
|
2023/1/11
|
|
||


















2022 News
|
2022/12/21 |
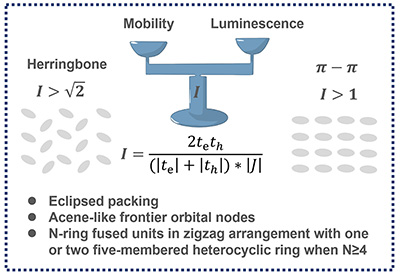
Congratulations to Qi Sun for the new paper accepted for publication in Adv. Optical Mater.! Integration of luminescence and carrier transport is of prime interest for optoelectronics, but full with challenges in organic semiconductors. Previously, it has been demonstrated that intermolecular charge transfer (ICT) in the herringbone H-aggregate can lend oscillator strength to the lowest-lying dark state if electron transfer (te) and hole transfer (th) integrals are in phase (same sign) and greater than √2J , where J is exciton coupling. In this work, both herringbone and π–π stackings are considered and a universal descriptor I = 2te th / [(|te| + |th|)·| J |] is proposed. It is found that for π–π stacking with I > 1 and for herringbone with I > √2, respectively, the lowest dark state could become bright through mixing ICT component. Accordingly, it is found that eclipsed packing and use of molecules with acene-like frontier molecular orbital profiles favor larger I value than the other packing patterns. Finally, with the help of the proposed descriptor, 13 high-mobility emissive unit cores from 32 fused ring molecules are effectively screened out. |
||
|
2022/11/25
|
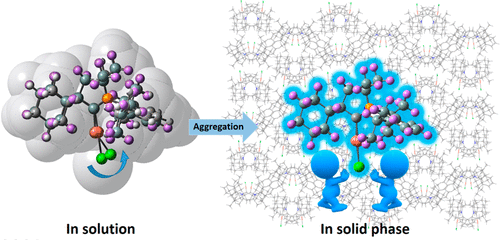
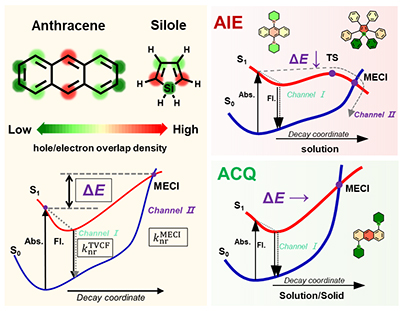
Congratulations to Ping-An Yin for the new paper accepted for publication in Aggregate! We computationally investigated the molecular aggregation effects on the excited state deactivation processes by considering both the direct vibrational relaxation and the S0/S1 surface crossing, that is, the minimum energy conical intersection (MECI). Taking classical AIEgens bis(piperidyl)anthracenes (BPAs) isomers and the substituted silole derivatives as examples, we show that the deformation ofMECI always occurs at the atom with greater hole/electron overlap. Besides, the energetic and structural changes of MECI caused by substituent has been investigated. We find that effective substituent such as the addition of the electron-donating groups, which can polarize the distribution of hole/electron density of molecules, will lead to the pyramidalization deformation of MECI occurring at the substituent position and simultaneously reduce the required energy to reach MECI. And MECI is sterically restricted by the surrounding molecules in solid phase, which remarkably hinders the non-radiative decay through surface crossing. Through quantitative computational assessments of the fluorescence quantum efficiency for both solution and solid phases, we elucidate the role of MECI and its dependence on the substitutions through pyramidalization deformation, which give rise to the aggregation-induced emission (AIE) phenomenon for 9,10-BPA, to aggregation-enhance emission (AEE) behavior for 1,4-BPA, and to conventional aggregation-caused quenching (ACQ) for 1,5-BPA. We further verify such mechanism for siloles, for which we found that the substitutions do not change the AIE behavior. Our findings render a general molecular design approach to manipulating the aggregation effect for optical emission.
|
||
|
2022/10/11
|
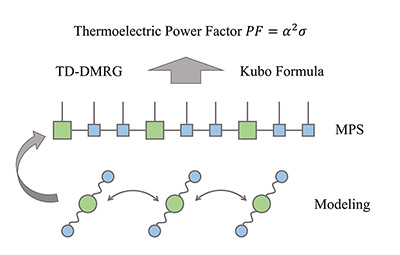
Congratulations to Yufei Ge for his new paper has been accepted for publication in JCTC! Organic/polymeric materials are of emerging importance for thermoelectric conversion. The soft nature of these materials implies strong electron–phonon coupling, often leading to carrier localization. This poses great challenges for the conventional Boltzmann transport description based on relaxation time approximation and band structure calculations. In this work, combining the Kubo formula with the finite-temperature time-dependent density matrix renormalization group (FT-TD-DMRG) in the grand canonical ensemble, we developed a nearly exact algorithm to calculate the thermoelectric power factor PF = α2 σ, where α is the Seebeck coefficient and σ is the electrical conductivity, and apply the algorithm to Holstein Hamiltonian with electron–phonon coupling to model organic materials. Our algorithm can provide a unified description covering the weak coupling limit described by the bandlike Boltzmann transport to the strong coupling hopping limit.
|
||
|
2022/10/07
|
|
||
|
2022/09/30 |
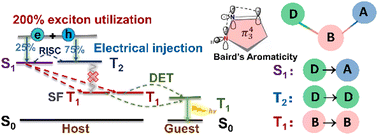
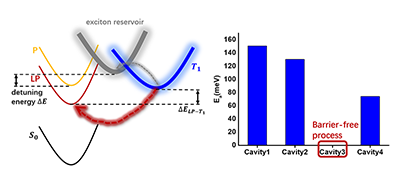
Congratulations to Dr Bin Zhang for the new paper published in JPCL! The lower polariton (LP) can reduce the energy barrier of the reverse intersystem crossing (rISC) process from T1 to harvest triplet energy for fluorescence. Based on a Tavis−Cummings model including both singlet and triplet excitons, both coupled with quantized photons, we derive here a comprehensive rISC rate formalism. We found that the latter consists of three contributions: the one originated from spin−orbit coupling as first obtained by Martinez-Martinez et al. (J. Chem. Phys. 2019, 151, 054106), the one from light−matter coupling of Ou et al. (J. Am. Chem. Soc. 2021, 143, 17786), and the cross-term first reported here. We apply the formalism to investigate the experimentally observed barrier-free rISC (BFrISC) process in cavity devices with DABNA-2 molecular thin film. We found it can be attributed to the detuning effect. The rISC rates can be increased by orders of magnitude through changing the detuning energy to realize the BFrISC process. In addition, the BFrISC rates exhibit a maximum as a function of the incident angle and the doping concentration. The formalism provides a solid ground for molecular design toward highly efficient cavity-promoted light-emitting materials.
|
||
|
2022/09/08 Congratulations on publication of the book "Density Matrix Renormalization Group (DMRG)-based Approaches in Computational Chemistry" (ISBN: 978-0-323-85694-2) |
Density Matrix Renormalization Group (DMRG)-based Approaches in Computational Chemistry outlines important theories and algorithms of DMRG-based approaches and explores their use in computational chemistry. Beginning with an introduction to DMRG and DMRG-based approaches, the book goes on to discuss the key theories and applications of DMRG, from DMRG for semi-empirical and ab-initio quantum chemistry, to DMRG in embedded environments, frequency spaces and quantum dynamics. Drawing on the experience of its expert authors, sections detail recent ideas and key developments, providing an up-to-date view of current developments in the field for students and researchers in quantum chemistry.
|
||
|
2022/08/03
|
|||
|
2022/07/24
|
帅志刚教授课题组招生和招聘启事 帅志刚教授在香港中文大学(深圳)建立课题组,从事理论化学与功能材料计算模拟研究。近几年的研究方向及代表作如下:
课题组开发的“分子材料性能”计算软件MOMAP已经成功地得到了商业化应用,用户包括海内外130多个课题组和有机发光材料公司的使用。课题组网页:http://www/shuaigroup.net/ . MOMAP软件页面: http://www.momap.net.cn 。 现招收博士研究生(含硕博生和博士生), 常年开放申请,对以上研究方向感兴趣的本科生或硕士研究生,请递交申请:http://gs.cuhk.edu.cn。春秋两次入学。如获录取,报道时需提供托福或雅思考试成绩,英语授课。 课题组长期招收博士后和研究助理,直接发邮件申请: |
||
|
2022/07/11
|
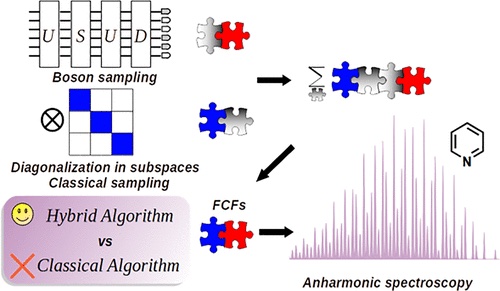
Congratulations to Yuanheng Wang for the new paper published in JPCL! Using a photonic quantum computer for boson sampling has demonstrated a tremendous advantage over classical supercomputers. It is highly desirable to develop boson sampling algorithms for realistic scientific problems. In this work, we propose a hybrid quantum-classical sampling (HQCS) algorithm to calculate the optical spectrum for complex molecules considering Duschinsky rotation effects and anharmonicity. The classical sum-over-states method for this problem has a computational complexity that exponentially increases with system size. The HQCS algorithm creates an intermediate harmonic potential energy surface (PES) to bridge the initial and final PESs. The magnitude and sign of the overlap between the initial and the intermediate state are estimated by boson sampling and classical algorithms, respectively. The overlap between the intermediate and the final state is efficiently evaluated by classical algorithms. The feasibility of HQCS is demonstrated in calculations of the emission spectrum of a Morse model as well as the pyridine molecule. |
||
|
2022/05/20
|
Congratulations to Weitang Li!
Congratulations to Weitang Li for completing his PhD Dissertation Defense!
|
||
|
2022/05/20
|
Congratulations to Shiyun Lin!
Congratulations to Shiyun Lin for completing her PhD Dissertation Defense!
|
||
|
2022/04/26
|
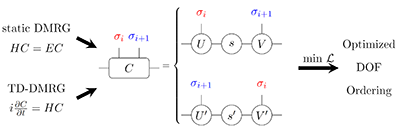
Congratulations to Weitang Li for the new paper published in J. of Physics: Condensed Matter! Density matrix renormalization group (DMRG) and its time-dependent variants have found widespread applications in quantum chemistry, including ab initio electronic structure of complex bio-molecules, spectroscopy for molecular aggregates, and charge transport in bulk organic semiconductors. The underlying wavefunction ansatz for DMRG, matrix product state (MPS), requires mapping degrees of freedom (DOF) into a one-dimensional topology. DOF ordering becomes a crucial factor for DMRG accuracy. In this work, we propose swapping neighboring DOFs during the DMRG sweeps for DOF ordering, which we term ‘on the fly swapping’ (OFS) algorithm.We show that OFS is universal for both static and time-dependent DMRG with minimum computational overhead. Examples are given for one dimensional antiferromagnetic Heisenberg model, ab initio electronic structure of N2 molecule, and the S1/S2 internal conversion dynamics of pyrazine molecule. It is found that OFS can indeed improve accuracy by finding better DOF ordering in all cases. |
||
|
2022/03/11
|
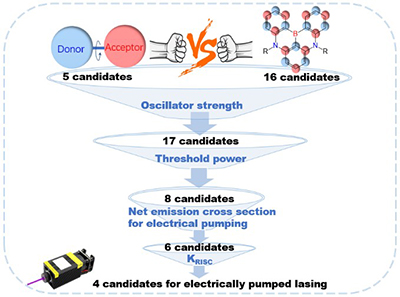
Congratulations to Shiyun Lin for the new paper published in ACS Materials Letters! Thermally activated delayed fluorescence (TADF) materials are competitive candidates toward electrically pumped organic lasing, because of its ability to suppress triplet accumulations by reverse intersystem crossing (RISC), especially, the multiresonance TADF (MR-TADF) compounds featuring narrow-band emission and high photoluminescence quantum yields. The goal of this work is to theoretically screen out promising electrically pumped organic laser compounds over both MR-TADF and conventional TADF molecules. We calculate the photophysical parameters over 21 organic TADF molecules to determine if the electrically pumped lasing criteria can be met, i.e., no substantial absorption/annihilation processes caused by excitons and polarons near the S1 emission wavelength. The selection criteria include large oscillator strength of S1, large net emission cross-section, long S1 lifetime, and large reverse intersystem crossing rate. We are able to conclude that DABNA-2, m-Cz-BNCz, ADBNA-Me-Mes, and ADBNA-Me-Tips MR-TADF molecules are prospective candidates for electrically pumped lasing based on our theoretical protocol, and we believe this work would immediately benefit this field with better and more efficient molecular design of TADF gain materials. |







2021 News
|
2021/11/09
|
Congratulations to Dr Jiajun Ren for the new paper published in CJCP! We propose a method for calculating the nonradiative decay rates for polyatomic molecules including anharmonic effects of the potential energy surface (PES) in the Franck-Condon region. The method combines the n-mode representation method to construct the ab initio PES and the nearly exact time-dependent density matrix renormalization group method (TD-DMRG) to simulate quantum dynamics. In addition, in the framework of TD-DMRG, we further develop an algorithm to calculate the final-state-resolved rate coefficient which is very useful to analyze the contribution from each vibrational mode to the transition process. We use this method to study the internal conversion (IC) process of azulene after taking into account the anharmonicity of the ground state PES. The results show that even for this semi-rigid molecule, the intramode anharmonicity enhances the IC rate significantly, and after considering the two-mode coupling effect, the rate increases even further. The reason is that the anharmonicity enables the C−H vibrations to receive electronic energy while C−H vibrations do not contribute on the harmonic PES as the Huang-Rhys factor is close to 0. |
||
|
2021/10/20
|
Congratulations to Dr Qi Ou for the new paper published in JACS! Polaritons are hybrid light−matter states formed via strong coupling between excitons and photons inside a microcavity, leading to upper and lower polariton (LP) bands splitting from the exciton. The LP has been applied to reduce the energy barrier of the reverse intersystem crossing (rISC) process from T1, harvesting triplet energy for fluorescence through thermally activated delayed fluorescence. The spin−orbit coupling between T1 and the excitonic part of the LP was considered as the origin for such an rISC transition. Here we propose a mechanism, namely, rISC promoted by the light−matter coupling (LMC) between T1 and the photonic part of LP, which is originated from the ISCinduced transition dipole moment of T1. This mechanism was excluded in previous studies. Our calculations demonstrate that the experimentally observed enhancement to the rISC process of the erythrosine B molecule can be effectively promoted by the LMC between T1 and a photon. The proposed mechanism would substantially broaden the scope of the molecular design toward highly efficient cavity-promoted light-emitting materials and immediately benefit the illumination of related experimental phenomena. |
||
|
2021/09/28
|
Congratulations to Dr Huili Ma for the 2019 JACS paper being one of the most highly cited publications in JACS for the period 2018-2019 ! |
||
|
2021/09/26
|
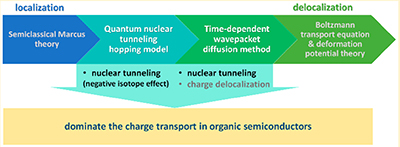
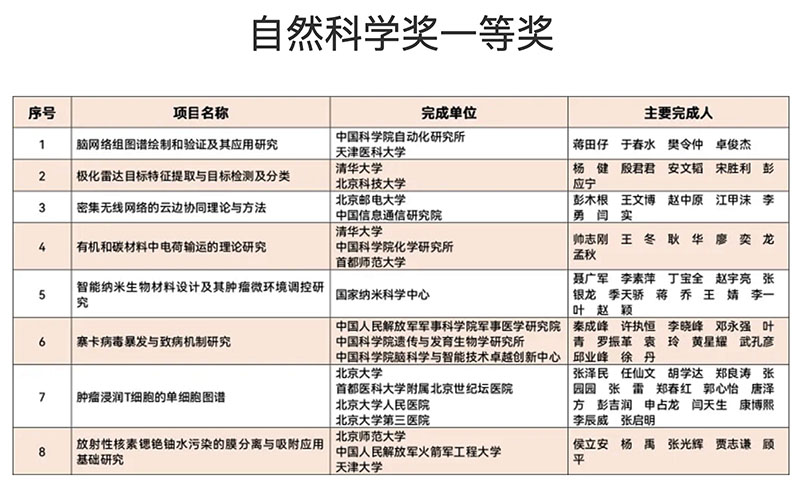
帅志刚教授主持的项目获2020年度北京市自然科学奖一等奖近日,2020年度北京市科学技术奖项目奖评审结果出炉,帅志刚教授主持的 “有机和碳材料中电荷输运的理论研究” 项目获2020年度北京市自然科学奖一等奖,获奖单位为清华大学、中国科学院化学研究所和首都师范大学。 新型有机和碳基材料中的电荷传输机理一直存在疑惑。2000年诺贝尔化学奖(导电高分子)的颁奖公告,采用Marcus电荷转移理论解释有机/高分子材料的导电现象,即局域化电荷(孤子或极化子)的热激活跳跃过程。但是从2006年到2009年,一批新的实验表明电导率在低温下并不消失,而是出现平台,与热激活模型矛盾。同时,基于Marcus理论所给出的电荷迁移率也明显低估了实验值,趋势也与实验不一致,从而阻碍分子设计。人们提出了包括一维强关联电子Luttinger液体在内的若干理论模型去解释实验结果,引发了激烈争论;而关于有机半导体中的载流子迁移率的同位素效应是正还是负的问,无论是实验还是理论都不明确,已经争论了50年!项目组考虑到碳原子的质量小,不仅仅会导致电子运动与原子振动紧密耦合,使得电子态出现局域化,同时还会表现出原子的量子涨落。据此,项目组推导了含原子量子涨落的电子转移速率公式,并提出了核隧穿驱动的电荷传输模型。该模型合理地解释了低温导电平台,同时指出同位素效应只能是负的:质量变大,量子涨落变小,从而驱动力变小。该模型被德国马普所所长Blom等人用于澄清高分子的电导之争,指出核隧穿驱动的极化子跳跃传输具有普适意义。同时,一批新的实验明确了同位素效应确实是负的,证实了项目组的理论预言。在此基础上,项目组发展考虑了电声子量子散射的迁移率计算方法,应用到碳基材料中,发现在石墨烯纳米带中电荷迁移率随纳米带宽度变化的尺寸效应,并指出新型碳材料石墨炔兼具高迁移率和直接带隙,是重要的电子材料;项目组还将计算方法应用到研究有机/高分子材料的热电转换,考察了化学掺杂对有机半导体能带结构、掺杂离子散射对电荷传输和热电转换的影响,计算得到的塞贝克系数得到多个实验组验证。 本项目得到国家自然科学基金委和国家科技部专项的支持。项目组将所发展的计算方法做成软件,得到来自哈佛、斯坦福等学者下载2600余次。2017年鸿之微(上海)科技有限公司将该程序商业化,目前国内外正式用户已达95家,包括11家海外用户。项目完成人应邀在第13届和第17届国际量子化学大会、第14届法国理论化学大会、第10届国际理论化学物理大会、以及国际合成金属大会上作全会特邀(Plenary)报告;由于在“电荷传输以及在理论化学领域的贡献”,获2012年中国化学会-阿克苏诺贝尔化学科学奖;由于在“激发态理论和2维材料领域开创性工作”,获2018年法国化学会“法-中讲座奖”;入选爱思唯尔2020年化学领域高被引作者。 |
||
|
2021/09/24
|
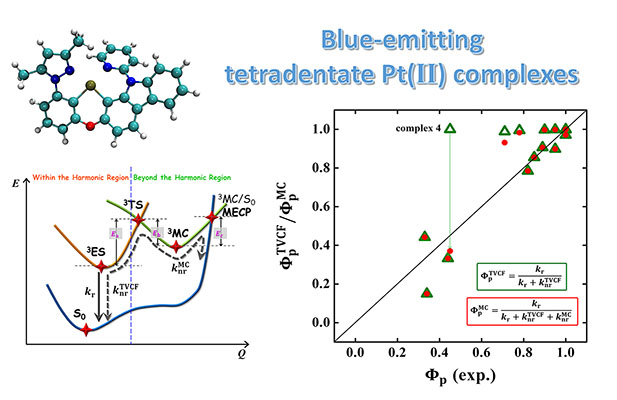
Congratulations to Dr Yu Wang for the new paper published in Materials Horizons! Phosphorescent organic light-emitting diodes (PhOLEDs) are leading candidates for displays or lighting technologies. Recently, blue phosphorescent tetradentate Pt(II) complexes have been attracting extensive attention due to their high phosphorescence quantum efficiency and numerous chemical structures on account of flexible ligand frames and modifications. Using quantum chemistry coupled with our thermal vibration correlation function (TVCF) formalism, we investigated the triplet excited state energy surface and the decay processes involving both direct vibrational relaxation and minimum energy crossing point (MECP) via the transition state (3TS) to the ground state (S0) for 16 recently experimentally reported blue-emitting tetradentate Pt(II) emitters containing fused 5/6/6 metallocycles. We found that (i) in most cases, the direct vibrational relaxation deactivations dominated the triplet non-radiative decay because either the 3TS is too high or the MECP is not reachable. Hence, results from the TVCF formalism agreed well with the experiments for the phosphorescence quantum efficiency; (ii) only when both 3TS and MECP are low, for instance, for PtON1-oMe, deactivations via MECP dominated the triplet non-radiative decay. |
||
|
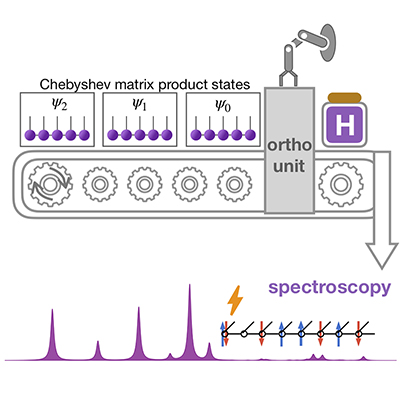
2021/09/23 |
Congratulations to Tong Jiang for the new paper published in J. Phys. Chem. Lett.! We propose a method to calculate the spectral functions of many-body systems by Chebyshev expansion in the framework of matrix product states coupled with canonical orthogonalization (coCheMPS). The canonical orthogonalization can improve the accuracy and efficiency significantly because the orthogonalized Chebyshev vectors can provide an ideal basis for constructing the effective Hamiltonian in which the exact recurrence relation can be retained. In addition, not only the spectral function but also the excited states and eigenenergies can be directly calculated, which is usually impossible for other MPS-based methods such as time-dependent formalism or correction vector. The remarkable accuracy and efficiency of coCheMPS over other methods are demonstrated by calculating the spectral functions of spin chain and ab initio hydrogen chain. For the first time we demonstrate that Chebyshev MPS can be used to deal with ab initio electronic Hamiltonian effectively. We emphasize the strength of coCheMPS to calculate the low excited states of systems with sparse discrete spectrum. We also caution the application for electron-phonon systems with dense density of states. |
||
|
2021/07/19
|
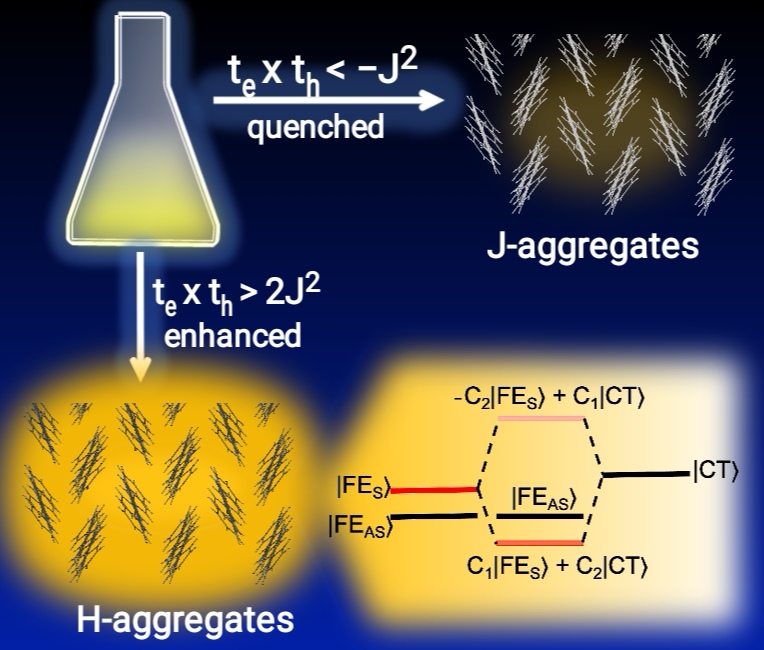
Congratulations to Qi Sun for the new paper published in Nano Letters! Luminescence in molecular aggregates can be quenched either by intermolecular charge transfer or by forming a dipole-forbidden lower Frenkel exciton in H-aggregate. Taking intermolecular charge transfer and excitonic coupling into a threestate model through localized diabatization, we demonstrate that the low-lying intermolecular charge-transfer state could couple with the upper bright Frenkel exciton to form dipole-allowed S1 that lies below the dark state, which accounts for the recent experimentally discovered strong luminescence in organic light-emitting transistors (OLETs) system with DPA and dNaAnt herringbone aggregates. The condition of forming such bright state is that the electron and hole transfer integrals, te and th, are of the same sign, and should be notably larger than the excitonic coupling (J), that is , te × th > 2J2. This theoretical finding not only rationalizes recent experiments but unravels an exciting scenario where strong luminescence and high charge mobilities become compatible, which is a preferable condition for both OLETs and electrically pumped lasing. |
||
|
2021/07/17
|
Congratulations to Weitang Li for the new paper published in Nature Communications!
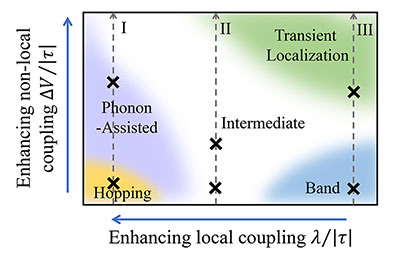
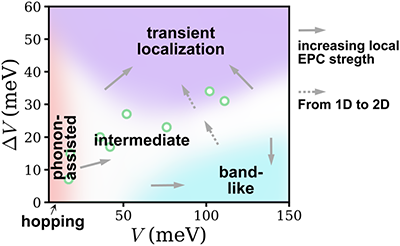
The nonlocal electron-phonon couplings in organic semiconductors responsible for the fluctuation of intermolecular transfer integrals has been the center of interest recently. Several irreconcilable scenarios coexist for the description of the nonlocal electron-phonon coupling, such as phonon-assisted transport, transient localization, and band-like transport. Through a nearly exact numerical study for the carrier mobility of the Holstein-Peierls model using the matrix product states approach, we locate the phonon-assisted transport, transient localization and band-like regimes as a function of the transfer integral (V) and the nonlocal electron-phonon couplings (ΔV), and their distinct transport behaviors are analyzed by carrier mobility, mean free path, optical conductivity and one-particle spectral function. We also identify an “intermediate regime” where none of the established pictures applies, and the generally perceived hopping regime is found to be at a very limited end in the proposed regime paradigm.
|
||
|
2021/07/17
|
Congratulations to Weitang Li for the new paper published in CJCU!
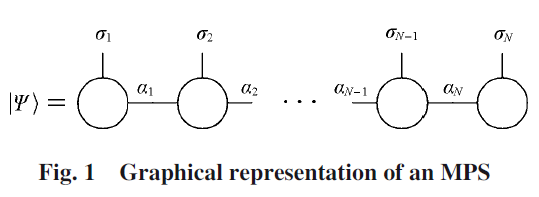
密度矩阵重正化群(DMRG)作为计算低维强关联体系强有力的方法为人熟知, 在量子化学电子结构计算中得到广泛应用. 最近几年, 含时密度矩阵重正化群(TD-DMRG)的理论取得较快发展, TD-DMRG逐渐成为复杂体系量子动力学理论模拟的重要新兴方法之一 . 本文综述了基于矩阵乘积态(MPS) 和矩阵乘积算符(MPO)的DMRG基本理论, 并重点介绍了若干最常见的TD-DMRG时间演化算法, 包括基于演化再压缩(P&C)的算法、基于含时变分原理(TDVP)的算法和时间步瞄准(TST)算法; 还对利用TD-DMRG模拟有限温体系的纯化(Purification)算法和最小纠缠典型量子热态(METTS)算法进行了介绍. 最后, 对近年TD-DMRG在复杂体系量子动力学中的应用进行了总结.
|
||
|
2021/06/17
|
Congratulations to Ran Liu for the new paper published in CCS Chemistry!
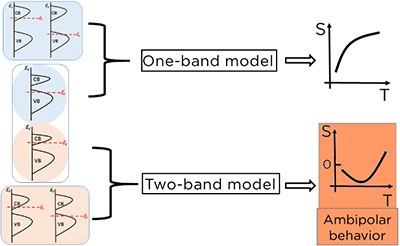
The Seebeck effect measures the electric potential built up in materials under a temperature gradient. For organic thermoelectric materials, the Seebeck coefficient shows more complicated temperature dependence than conventional systems, with both monotonic increases and nonmonotonic behavior, that is, first increasing and then decreasing. The mechanism behind the phenomenon is intriguing. Through first-principles calculations coupled with the Boltzmann transport equation, we demonstrate typical trends of the Seebeck coefficient with respect to temperature through band structure analysis. The bandgap and bandwidths of the valence band and conduction band jointly determine the effectiveness of thermal activation. Ineffective or effective thermal activation leads to a one- or a two-band transport behavior, respectively. Under the thermal-activation mechanism, Seebeck coefficient shows monotonic temperature dependence in a one-band model but nonmonotonic relationship in a two-band model. In particular, in the two-band model, Seebeck coefficient might show an ambipolar behavior, that is, its sign changes at high temperature.
|
||
|
2021/06/03
|
Congratulations to Yuanheng Wang for the new paper published in J. Chem. Phys.!
In this work, we propose a new method to calculate molecular nonradiative electronic relaxation rates based on the numerically exact timedependent density matrix renormalization group theory. This method could go beyond the existing frameworks under the harmonic approximation (HA) of the potential energy surface (PES) so that the anharmonic effect could be considered, which is of vital importance when the electronic energy gap is much larger than the vibrational frequency. We calculate the internal conversion (IC) rates in a two-mode model with Morse potential to investigate the validity of HA. We find that HA is unsatisfactory unless only the lowest several vibrational states of the lower electronic state are involved in the transition process when the adiabatic excitation energy is relatively low. As the excitation energy increases, HA first underestimates and then overestimates the IC rates when the excited state PES shifts toward the dissociative side of the ground state PES. On the contrary, HA slightly overestimates the IC rates when the excited state PES shifts toward the repulsive side. In both cases, a higher temperature enlarges the error of HA. As a real example to demonstrate the effectiveness and scalability of the method, we calculate the IC rates of azulene from S1 to S0 on the ab initio anharmonic PES approximated by the one-mode representation. The calculated IC rates of azulene under HA are consistent with the analytically exact results. The rates on the anharmonic PES are 30%–40% higher than the rates under HA.
|
||
|
2021/05/24
|
Congratulations to Lu Wang!
Congratulations to Lu Wang for completing her PhD Dissertation Defense!
|
||
|
2021/05/24
|
Congratulations to Guodong Wang!
Congratulations to Guodong Wang for completing his PhD Dissertation Defense!
|
||
|
2021/05/24
|
Congratulations to Sheng Huang!
Congratulations to Sheng Huang for completing his Master Dissertation Defense!
|
||
|
2021/03/28
|
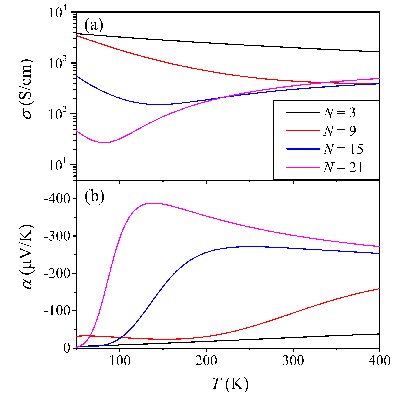
Congratulations to Yufei Ge on the published paper in Appl. Phys. Lett.! The Seebeck effect or thermopower relates the temperature gradient to the electric voltage drop. Seebeck coefficient a measures the transport entropy, which could either linearly increase with temperature T like metallic conducting or decrease as 1/T like semiconducting behavior. It could become more complicated in the temperature dependence for a number of disordered systems but still in a monotonic way. However, several recent experiments reported the “abnormal” non-monotonic temperature dependence of the Seebeck coefficient in doped conducting polymers, for instance, first increasing and then decreasing. Through a one-dimensional tight-binding model coupled with the Boltzmann transport equation, we investigate theoretically the doping effect for the Seebeck coefficient. We find that the abnormal behavior comes from multi bands’ contribution and a two-band model (conduction or valence band plus a narrow polaronic band) can address such an abnormal Seebeck effect, namely, if there exists (i) a small bandgap accessible for thermal activation between the two bands; and (ii) a large difference in the bandwidth between the polaronic band and the conduction band (or valence band), then the Seebeck coefficient increases with temperature first, then levels off, and finally drops down. |
||
|
2021/03/18
|
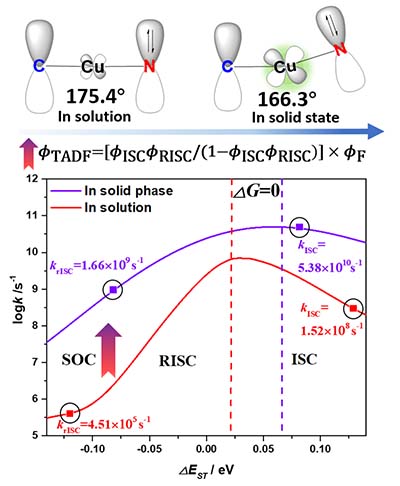
Congratulations to Shiyun Lin on the published paper in J. Phys. Chem. Lett.! The two-coordinate carbene−metal−amide complexes have attracted a great deal of attention due to their remarkable thermally activated delayed fluorescence (TADF) properties, giving them promise in organic light-emitting diode application. To reveal the inherent mechanism, we take CAAC−Cu(I)−Cz and CAAC−Au(I)−Cz as examples to investigate the photophysical properties in solution and solid phases by combining quantum mechanics/molecular mechanics approaches for the electronic structure and the thermal vibration correlation function formalism for the excited-state decay rates. We found that both intersystem crossing (ISC) and its reverse (rISC) are enhanced by 2−4 orders of magnitude upon aggregation, leading to highly efficient TADF, because (i) the metal proportion in the frontier molecular orbitals increases, leading to an enhanced spin−orbit coupling strength between S1 and T1, and (ii) the reaction barriers for ISC and rISC are much lower in solution than in aggregate phases through a decrease in energy gap ΔEST and an increase in the relative reorganization energy through bending the angle ∠C2−Cu−N1 for T1. We propose a pump−probe time-resolved infrared spectroscopy study to verify the mechanism. These findings can clarify the ongoing dispute over the understanding of the high TADF quantum efficiency for two-coordinate metal complexes. |
||
|
2021/03/15
|
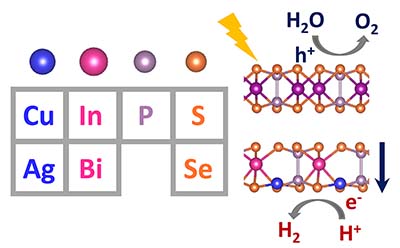
Congratulations to Sheng Huang on the published paper in J. Mater. Chem. A! Integration of ferroelectricity into van der Waals heterostructures offers additional opportunities to control over the properties and functionalities of heterostructures by switching the direction of polarization via an external electric field. To discover potential ferroelectric monolayers that exhibit out-of-plane electric polarizations, we screen a family of metal phosphorus trichalcogenides M1M2P2X6 with M1= Cu/Ag, M2 = In/Bi, X = S/Se using density functional theory calculations. We predict room-temperature ferroelectricity/ in CuInP2S6 and CuBiP2S6 monolayers with out-of-plane polarizations (Ps) of 0.59 μC cm-2 and 0.35 μC cm-2, respectively. The strong metal-chalcogenide M1-X bond in the two Cu and S-based systems is responsible for their high phase transition temperatures. The polarizations in ferroelectric monolayers can persist in van der Waals heterostructures, and band gaps as well as band alignment of the heterostructures can be tuned by switching the polarization direction. Finally, we demonstrate that both visible light absorption and type-II band alignment facilitating fast charge separation can be realized in CuInP2S6/Mn2P2S6 and CuInP2S6/Zn2P2Se6 ferroelectric heterostructures, which are promising for applications in photocatalytic water splitting. |
||
|
2021/03/15
|
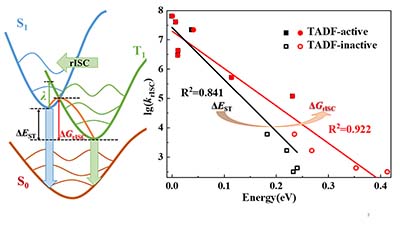
Congratulations to Lu Wang on the published paper in J. Phys. Chem. A! The thermally activated delayed fluorescence (TADF) phenomenon has attracted increasing attention because it can harvest 100% of the electro-pumped carriers to form singlet bound excited state for fluorescence. It is generally believed that the small energy gap between S1 and T1 (ΔEST) is essential for TADF to facilitate the reverse intersystem crossing (rISC). However, for a few donor-acceptor (D-A) organic compounds with small ΔEST, the TADF phenomenon is absent, indicating that ΔEST might not be a good molecular descriptor. Here, using our self-developed thermal vibration correlation function (TVCF) formalism in combination with quantum chemistry calculations, we revisit the key factors that dominate the TADF property for 11 D-A systems with small ΔEST. Based on our theoretical results in comparison to experiments, we conclude that the activation energy ΔG is a good molecular descriptor to characterize the TADF performance because a significantly better linear relationship is observed between ΔG and the rISC rate constant (krISC) compared to that between ΔEST and krISC. These findings provide deeper understanding of the TADF mechanism, shedding light on the molecular design of high-performance TADF materials. |
||
|
2021/03/15
|
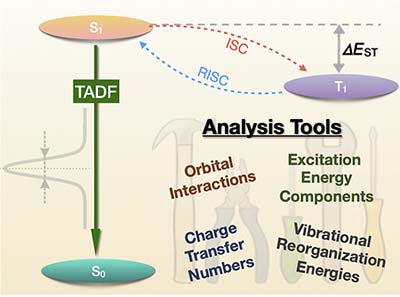
Congratulations to Dr Qi Ou on the published paper in J. Phys. Chem. Lett.! Recently, Wang and co-workers carried out frontier molecule orbital engineering in the design of m-Cz-BNCz, a thermally activated delayed fluorescence (TADF) molecule that emits pure green light at an external quantum efficiency of 27%. To further understand the underlying molecular design principles, we employed four advanced electronic structure analysis tools. First, an absolutely localized molecular orbitals (ALMO-) based analysis indicates an antibonding combination between the highest occupied molecular orbitals (HOMOs) of the donor 3,6-di-tert-butylcarbazole fragment and the acceptor BNCz fragment, which raises the HOMO energy and red-shifts the fluorescence emission wavelength. Second, excitation energy component analysis reveals that the S1−T1 gap is dominated by two-electron components of the excitation energies. Third, charge transfer number analysis, which is extended to use fragment-based Hirshfeld weights, indicates that the S1 and T1 excited states of m-Cz-BNCz (within time-dependent density functional theory) have notable charge transfer characters (27% for S1 and 12% for T1). This provides a balance between a small single-triplet gap and a substantial fluorescence intensity. Last, a vibrational reorganization energy analysis pinpoints the torsional motion between the BNCz and Cz moieties of m-Cz-BNCz as the source for its wider emission peak than that of p-Cz-BNCz. These four types of analyses are expected to be very valuable in the study and design of other TADF and functional dye molecules. |
||
|
2021/02/17
|
|
||
|
2021/02/14
|
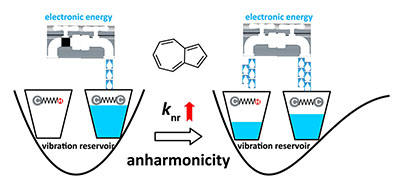
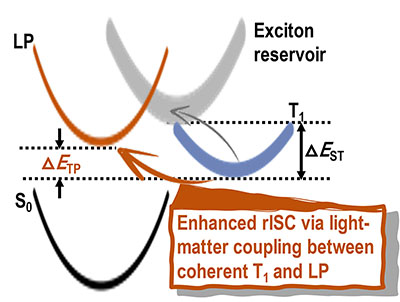
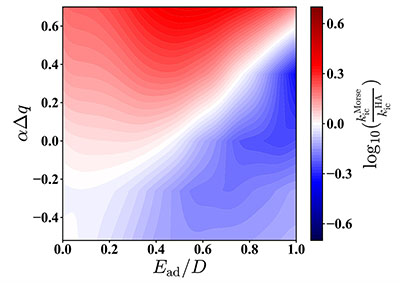
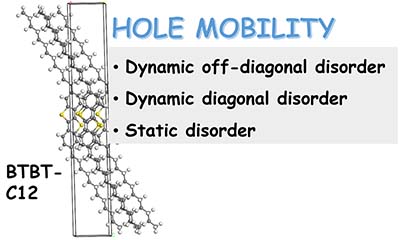
Congratulations to Dr Xingliang Peng on the published paper in Nanoscale! A multiscale approach by combining molecular dynamics simulations, quantum mechanics calculations and kinetic Monte-Carlo simulations has been applied to investigate the influence of dynamic and static disorder on the hole mobilities of four BTBT isomers. It is found that the dynamic disorder of transfer integral tends to decrease the mobility for quasi-1D (quasi one-dimensional) BTBT1 and BTBT4 isomers and increase the mobility for 2D (two-dimensional) BTBT2 and BTBT3 isomers, while the dynamic disorder of site energy tends to decrease mobility for all the four isomers; however, the reduction in 2D molecules is much less than that in quasi-1D molecules. Results show that trap defects could reduce the mobility for both the quasi-1D and 2D molecular structures significantly, even to several orders of magnitude.
|






2020 News
|
2020/12/10
|
|
||
|
[2020/9/23]
|
Congratulations to Dr Yu Wang on the published paper in J. Mater. Chem. A! It is generally perceived that a fast reverse intersystem crossing rate of T1 / S1 (krisc) is crucial for efficient organic thermally activated delayed fluorescence (TADF) emitters, the paper demonstrates the nonradiative decay rate of T1 / S0 (kTnr) for transition metal complexes that is even more important. |
||
|
[2020/9/16]
|
Congratulations to Dr Qi Ou on the published paper in Nature Communications ! Electrically pumped organic lasing is one of the most challenging issues in organic optoelectronics. The article present a systematic theoretical investigation to screen out electrical pumping lasing molecules over a wide range of organic materials. With the electronic structure information obtained from time-dependent density functional theory, multiple photophysical parameters of a set of optical pumping organic laser are calculated with MOMAP to judge whether the electrically pumped lasing conditions can be satisfied, namely, to avoid reabsorption from excitons and/or polarons, and the accumulation of triplet excitons. |
||
|
[2020/9/11]
|
|
||
|
[2020/7/3]
|
|
||
|
[2020/6/21]
|
Congratulations to Weitang Li for the new paper published in J. Phys. Chem. Lett.! Congratulations to Weitang Li for his new paper on Finite-Temperature TD-DMRG for the Carrier Mobility of Organic Semiconductors has been published in J. Phys. Chem. Lett. |
||
|
[2020/4/30]
|
Congratulations to Tong Jiang for the new paper published in J. Phys. Chem. Lett.! Congratulations to Tong Jiang for his new paper on Finite Temperature Dynamical Density Matrix Renormalization Group for Spectroscopy in Frequency Domain has been published in J. Phys. Chem. Lett. |
||
|
[2020/4/21]
|
Congratulations to Yajing Sun for the new paper published in ChemPhysChem! Congratulations to Yajing Sun for his new paper on Computational Study on the Charge Transport and Optical Spectra of Anthracene Derivatives in Aggregates has been published in ChemPhysChem. |
||
|
[2020/1/15]
|
Congratulations to Weitang Li for the new paper published in J. Chem. Phys.! Congratulations to Weitang Li for his new paper on Numerical assessment for accuracy and GPU acceleration of TD-DMRG time evolution schemes has been published in Inorg. Chem. |





2019 News
|
[2019/12/3]
|
Congratulations to Shiyun Lin for the new paper published in Inorg. Chem.! Congratulations to Shiyun Lin for his new paper on Strong Solid-State Fluorescence Induced by Restriction of the Coordinate Bond Bending in Two-Coordinate Copper(I)–Carbene Complexes has been published in Inorg. Chem.
|
|
[2019/12/3]
|
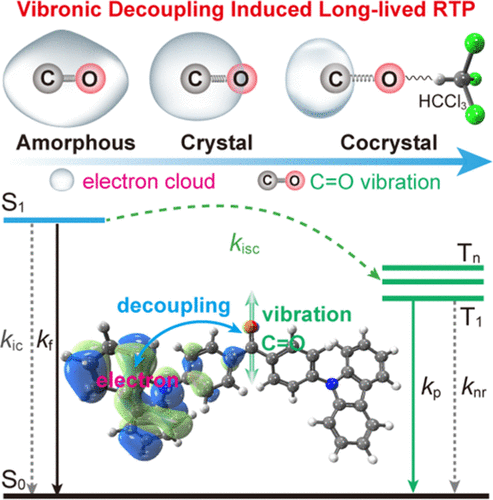
Congratulations to Huili Ma for the new paper published in Journal of Physical Chemistry Letters! Congratulations to Huili Ma for his new paper on Hydrogen Bonding-Induced Morphology Dependence of Long-Lived Organic Room-Temperature Phosphorescence has been published in Journal of Physical Chemistry Letters. |
|
[2019/09/01]
|
Welcome Dr Xingliang Peng, Dr Qi Ou, Dr Yu Wang, Mr Hengrui Yang and Mr Yufei Ge to join the Shuaigroup!
|
|
[2019/05/29]
|
Congratulations to Zhen Wang!
Congratulations to Zhen Wang for completing his PhD Dissertation Defense!
|
|
[2019/05/29]
|
Congratulations to Jahanzeb Khan!
Congratulations to Jahanzeb Khan for completing his PhD Dissertation Defense!
|
|
[2019/05/29]
|
Congratulations to Yajing Sun!
Congratulations to Yajing Sun for completing her PhD Dissertation Defense!
|
|
[2019/05/29]
|
Congratulations to Jiajun Ren!
Congratulations to Jiajun Ren for completing his PhD Dissertation Defense!
|
|
[2019/05/20]
|
Congratulations to Yunpeng Liu for the new paper published in Journal of Physical Chemistry Letters! Congratulations to Yunpeng Liu for his new paper on boosting the Seebeck coefficient for organic coordination polymers has been published in Journal of Physical Chemistry Letters. |
|
[2019/05/18]
|
Congratulations to Yajing Sun for the new paper published in Journal of Physical Chemistry C! Congratulations to Yajing Sun for her new paper on the role of phonon–electron coupling in reducing lattice thermal conductivity has been published in Journal of Physical Chemistry C. |
|
[2019/05/18]
|
Congratulations to Wen Shi for the new paper published in Advanced Electronic Materials! Congratulations to Wen Shi for his new paper on high-performance thermoelectric materials has been published in Advanced Electronic Materials.
|
|
[2019/03/29]
|
Congratulations to Yuqian Jiang for the new paper published in Journal of Chemical Theory Computation! Congratulations to Yuqian Jiang for her new paper on the charge transport in organic semiconductors has been published in Journal of Chemical Theory Computation. |
|
[2019/02/10]
|
Congratulations to Tian Zhang for the new paper published in Journal of Materials Chemistry C! Congratulations to Tian Zhang for her new paper on organic mechano-responsive luminescent materials has been published in Journal of Materials Chemistry C. |
|
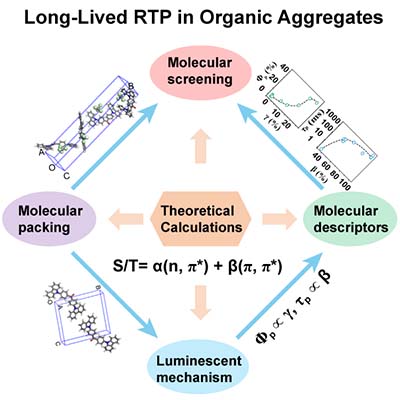
[2019/01/18]
|
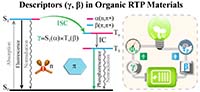
课题组在有机室温磷光材料理论设计方面取得重要进展 由于纯有机分子的旋轨耦合比较弱,长期以来有机材料室温下没有磷光。最近,一系列实验发现在固相下纯有机化合物也会表现出高效室温磷光,但其发光效率和寿命不可兼得。在帅志刚和彭谦的指导下,博士后马会利等人提出了一对分子描述符来分别表征磷光效率和寿命。由羰基和π-共轭片段组成的典型RTP体系,其激发态可以视为n→π*跃迁和π→π*跃迁两组分的组合。他们基于光物理原理,特别是El-Sayed规则,引入了一对分子描述符 (γ, β), 其数值的大小与固相下单/三线激发态的(n, π*)和(π, π*)跃迁成分有关。结合量子力学/分子力学(QM/MM)方法,他们揭示了分子描述符 (γ, β) 与磷光效率和寿命以及旋轨耦合之间的关系。他们提出,大的γ和β值有利于有机材料中强的、长寿命的RTP。该研究以题为“Efficient and Long-lived Room Temperature Organic Phosphorescence: Theoretical Descriptors for Molecular Designs”发表于Journal of the American Chemical Society,2019,141,1010-1015。 Read more... |